|
|
GENETISCHE AFWIJKINGEN BIJ DE HOND (CANIS FAMILIARIS) |
Genetic diseases of the dog (Canis familiaris) and their molecular genetic and biochemical characterisation
Prof.Dr. L.J.Peelman, Prof. Dr. A. Van Zeveren, Dr. F. Coopman, Prof. Dr. Y. Bouquet
Centrum voor Moleculaire en Biochemische Genetica van de Huisdieren, Faculteit Diergeneeskunde, Universiteit Gent, Heidestraat 19, B-9820 Merelbeke
|
SAMENVATTING
Een overzicht wordt gegeven van de genetische afwijkingen bij de hond waarvan het oorzakelijk genetisch defect op moleculair genetisch niveau is gekarakteriseerd en/of waarvan het deficiënte eiwit aan de hand van biochemische technieken is geďdentificeerd. Een aantal van de talrijke afwijkingen waarvoor aanwijzingen bestaan die duiden op een genetische oorsprong, maar waarvoor (nog) geen deficiënte factor is gekend, worden eveneens opgesomd, maar niet in detail besproken. Voor elk van de genetische afwijkingen met gekende deficiëntie wordt kort de pathologie, de overerving en de identiteit van de defecte factor toegelicht. ABSTRACT An overview is presented of those genetic disorders in dogs of which the molecular defect is known and/or the deficient factor has been determined with biochemical methods. A list of defects with a probable genetic basis but for which the deficient factor has not (yet) been determined is also included. For each of the disorders a short description of the pathology, the inheritance pattern and the nature of the defective factor is given. KEY WORDS: Dog - Genetic defects - Identification
INLEIDING
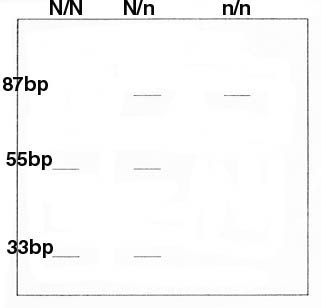
Een wijziging in de DNA-samenstelling van een gen (mutatie) kan aanleiding geven tot de aanmaak van een aberrant enzyme of de oorzaak zijn van te hoge of te lage enzyme concentraties. Een mutatie die aanleiding geeft tot een defect eiwit hoeft nog niet noodzakelijk klinische symptomen te veroorzaken. Elk individu heeft immers twee kopijen van elk gen (uitzondering hierop vormen de genen gelegen op de geslachtschromosomen bij mannelijke zoogdieren). Bij defect van één van de twee homologe genen produceert het normaal gen mogelijks voldoende functioneel eiwit. In dat geval is het defect gen (allel) recessief tegenover het normaal gen (allel), dat dominant wordt genoemd. In de regel zijn defecten in enzymen recessief, terwijl die in structurele eiwitten dominant zijn. Wanneer een defect allel recessief is tegenover het normale allel, dan zal de afwijking enkel tot uiting komen wanneer een dier twee defecte allelen heeft, en dus homozygoot is voor de mutatie (tenzij voor een X-gebonden kenmerk bij een _ (XY) individu). Hierbij dient opgemerkt te worden dat een heterozygoot dier in dit geval weliswaar geen klinische symptomen zal ontwikkelen, maar wel drager is van de genetische afwijking en deze ook aan 50 % van zijn nakomelingen zal doorgeven. Indien de paringspartner eveneens heterozygoot is, dan verwacht men 25 % aangetaste nakomelingen, naast 50 % klinisch normale dragers en 25 % fokzuivere normale individuen. Het is dan ook heel belangrijk voor de fokkerij om te kunnen beroep doen op een techniek, hetzij moleculair genetisch hetzij biochemisch, waarmee heterozygoten van homozygoten kunnen onderscheiden worden binnen de groep van klinisch normale dieren. Bovendien is het interessant dieren, homozygoot voor een aandoening die zich pas op latere leeftijd manifesteert (bv. progressieve retina atrofie), vroegtijdig als dusdanig te kunnen herkennen, en ze vervolgens preventief uit de fokkerij te weren. Aangezien in eenzelfde gen op verschillende plaatsen mutaties kunnen optreden is het aantal mogelijke genetische afwijkingen in theorie groter dan het aantal genen in een organisme. Het aantal genen per haploďd genoom (n=39 chromosomen bij de hond) van hogere zoogdieren wordt geschat tussen de 70 000 en 100 000. De meeste schadelijke mutaties en eruit voortvloeiende afwijkingen zijn evenwel dominant lethaal in een vroeg stadium van de embryonale ontwikkeling en kunnen aldus niet doorgegeven worden in een populatie. Een bijkomende manier om de prevalentie van genetische afwijkingen te beperken ligt in het opstellen van een verantwoord fokschema waarbij rekening moet gehouden worden met de genetische achtergrond van de fokdieren (studie van de stamboom). Dit is des te waardevoller naarmate het effectief aantal dieren binnen een ras kleiner is. Nogal wat rassen hebben bovendien een nauwe genetische basis, zodat het vermijden van een te grote inteeltbelasting bij de voortgebrachte pups essentieel is. Indien er twijfels bestaan over de identiteit of de herkomst van een bepaald fokdier, of gewoon ter controle van de voorgehouden origines, is het heel belangrijk te kunnen beschikken over een methode die individuele dieren als dusdanig ondubbelzinnig herkenbaar maakt. Een elegante techniek in dit verband is DNA fingerprinting, waarvan bepaalde varianten zijn ontwikkeld die worden toegepast bij de identiteitsbepaling en ouderschapscontrole in hondenpopulaties. Hierbij wordt gebruik gemaakt van PCR (Polymerase Chain Reaction) en van zgn. microsatellieten, waarvan er reeds tientallen beschreven zijn (Mellersh et al., 1994) Sommige van de hierna besproken genetische afwijkingen worden aangetroffen in het merendeel van de rassen, terwijl andere specifiek zijn voor een welbepaald ras. Indien mogelijk wordt telkens aangegeven in welke rassen de afwijking het meest wordt aangetroffen. GENETISCHE AFWIJKINGEN MET GEKEND MOLECULAIR DEFECT Basisprincipe van de moleculaire diagnosen Fucosidosis, haemofilie B, Krabbeziekte, mucopolysaccharidosis I (MPS I), musculaire dystrofie, pyruvaatkinase deficiëntie van erythrocyten, rod/cone dysplasia I, tremor, X-gebonden Severe Combined Immunodeficiency (SCID) en X-gebonden nephritis zijn genetische afwijkingen waarvan het defect op moleculair genetisch niveau is opgehelderd (Tabel 1) en waarvoor in het laboratorium methodes zijn ontwikkeld welke routinematig kunnen gebruikt worden om de mutatie op te sporen. Aldus kunnen ook de heterozygote dragers welke geen klinische symptomen vertonen geďdentificeerd worden. De moleculaire techniek bestaat erin dat vooreerst een klein stukje van het gen waarin de mutatie gelegen is door middel van de polymerase kettingreactie (PCR) wordt vermeerderd. Daarna wordt de geamplificeerde DNA-strook met een heel specifiek restrictie enzyme geknipt. Er zijn twee mogelijkheden: ofwel vernietigde de mutatie de herkenningsplaats van het restrictie enzyme in de DNA-strook, ofwel creeërde ze er daarentegen een in het defect gen, terwijl ze niet voorkomt in het normaal gen. In het eerste geval wordt het normaal gen geknipt en het defect gen niet, in het tweede geval gebeurt het omgekeerde. Als voorbeeld wordt de techniek voor detectie van MPS I (Menon et al., 1992) gegeven in figuur 1. D.m.v. PCR wordt een fragmentje van 87 baseparen (bp) van het a-L-iduronidasegen vermeerderd. Dit fragmentje wordt vervolgens behandeld met een restrictie enzyme waarvan de herkenningsplaats GGTGAN is. Dit enzyme knipt het 87 bp fragment afkomstig van het normale allel in twee kleinere stukken, zijnde 55 bp en 32 bp. De mutatie verantwoordelijk voor MPS I vernietigt echter de herkenningsplaats door de transitie van G (guanine) naar A (adenine) (GATGAN), zodat het 87 bp fragment van het mutante allel niet wordt geknipt. De bekomen fragmentjes kunnen zichtbaar gemaakt worden door elektroforese in een agarosegel. Het elektroforesepatroon van homozygoot normale dieren levert twee bandjes op van resp. 55 en 32 bp. Het patroon van heterozygote dieren vertoont drie bandjes, namelijk van 87, 55 en 32 bp, terwijl dat van homozygoot mutante dieren slechts één bandje geeft van 87 bp.
Fig.1. Moleculair diagnostiche test voor mucopolyschacharidosis. Schematische voorstelling van de allelen na scheiding in een agarosegel N-normaal allel, n=mutant allel bp=baseparen
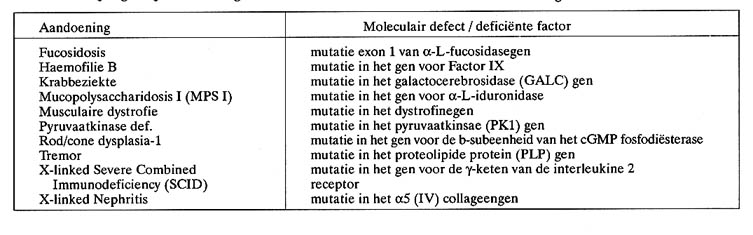
Tabel 1. Genetische afwijkingen bij de hond met een gekend moleculair defect en waarvoor een moleculaire diagnostische test werd ontwikkeld. De DNA-testen voor de overige afwijkingen samengebracht in tabel 1 gebeuren volgens een analoog basisconcept. Deze diagnostische DNA-testen kunnen routinematig worden uitgevoerd in het Centrum voor Moleculaire en Biochemische Genetica van de Huisdieren. Voor meer details over de praktische uitvoering kan kontakt opgenomen worden met één van de auteurs van deze publicatie. Fucosidosis Klinische symptomen en voorkomen: fucosidosis werd beschreven voor de Springer Spaniel (Barker et al., 1988) en wordt gekenmerkt door de afwezigheid van actief a-L-fucosidase met een opstapeling van glycoconjugaten in de lysosomen tot gevolg. De meeste symptomen, o.a. motorische storingen, worden veroorzaakt door beschadigingen in het centraal zenuwstelsel. De ziekte wordt eveneens gekenmerkt door groei vertraging en visceromegalie. De afwijking kan gecorrigeerd worden door beenmergtransplantatie of via gentherapie waarbij d.m.v een retrovirale vector het functionele a-L-fucosidasegen wordt ingebracht (Taylor et al., 1992). Overerving en karakteristieken van de defecte factor: de afwijking wordt veroorzaakt door een deletiemutatie van 14 baseparen in het eerste exon (= EXpressed regiON = een van de delen van het gen dat wordt vertaald in aminozuren) van het gen voor a-L-fucosidase (Skelly et al., 1996) en erft autosomaal (= niet geslachtsgebonden) recessief over. De mutatie veroorzaakt tevens een leesraamverschuiving waardoor een niet functioneel product van slechts 152 aminozuren wordt aangemaakt. Haemofilie B Klinische symptomen en voorkomen: haemofilie B wordt veroorzaakt door een defect in bloedstollingsfactor IX en werd aangetroffen in o.a. Cairn Terrier, Sint-Bernard, Franse Bulldog, Duitse Herder en Cocker Spaniel. De hemorragie is in haemofilie B meestal milder dan in het type A, is net als A afhankelijk van de grootte van de hond en ernstiger in de grotere rassen. Haemofilie B kan in tegenstelling met A (zie verder) gecorrigeerd worden door toediening van normaal serum (Brinkhous et al., 1973). Overerving en moleculair defect: zowel haemofilie A als B zijn X-gebonden maar worden niet gecontroleerd door hetzelfde locus. Populatiestudies hebben aangetoond dat de loci voor A en B ver (ongeveer 50 cM) van elkaar gelegen zijn. Haemofilie B was één van de eerste genetische afwijkingen bij huisdieren die werd opgehelderd met moleculair genetische technieken (Evans et al., 1989). Het defect wordt veroorzaakt door een transitiemutatie in positie 1477 van het gen voor bloedstollingsfactor IX. Het normale allel heeft in positie 1477 een guanine (G) en het mutante allel een adenine (A). Het mutante allel codeert aldus voor een eiwit met een glutaminezuur in positie 379 terwijl het normale eiwit op deze plaats een glycine bevat. De vervanging van glycine door glutaminezuur op deze plaats heeft nefaste gevolgen op de structuur en functie van het factor IX eiwit. Krabbe ziekte (globoid cell leucodystrofie, galactocerebrosidase def.) Klinische symptomen en voorkomen: Krabbeziekte werd o.a. aangetroffen in West Highland White Terrier, Cairn Terrier, Poedel, Bluetick Hound, Beagle en Dalmatiërs (Johnson et al., 1975). De ziekte is progressief en kent een snel verloop. De symptomen worden zichtbaar na 4 tot 12 weken. Coördinatie en beweging zijn gestoord. Tremor en zwakte van de ledematen verschijnen eerst, gevolgd door spieratrofie en degeneratie van het zenuwstelsel. Myelinescheden van perifere zenuwen en witte materie van het centraal zenuwstelsel degenereren en na verloop wordt de witte materie vervangen door globoďde cellen. Bij een leeftijd van 8 ŕ 9 maanden zijn de symptomen zo ernstig dat de dieren meestal worden geëuthanaseerd (Fletcher en Kurtz, 1972).
Overerving en moleculair defect: de ziekte erft over als een autosomaal recessief kenmerk en wordt veroorzaakt door een transitiemutatie (een adenine wordt vervangen door een cytosine) in positie 473 van het galactocerebrosidasegen (GALC) (Victoria et al., 1996). Het galactocerebrosidase hydrolyseert galactolipiden waaronder het galactocerebroside (galatosylceramide), het hoofdlipide in myeline en een belangrijk bestanddeel van nier- en epitheelcellen van de darm, en psychosine (Wenger en Chen, 1993). Het normale allel codeert voor een eiwit met een tyrosine (hydrofoob) in positie 158 terwijl het mutante allel een serine (hydrofiel) in deze positie heeft. Deze mutatie verlaagt de aktiviteit van het enzyme drastisch, zoals werd vastgesteld in homozygoot mutante dieren. Een tweede transitiemutatie (cytosine 6 thymine) in positie 1915 werd aangetroffen maar deze mutatie lijkt geen invloed te hebben op de activiteit van het galactocerebrosidase (Victoria et al., 1996).
Mucopolysaccharidosis I (MPS I) Klinische symptomen en voorkomen: MPS I omvat een groep van afwijkingen veroorzaakt door een deficiëntie in het lysosomale enzyme a-L-iduronidase en gebrek aan opstapeling van de substraten (dermataansulfaat en heparaansulfaat) in de lysozomen. Gewrichtsstijfheid, vertroebeling van het hoornvlies, vergroting van organen en cardiovasculaire aandoeningen worden in de meeste gevallen aangetroffen. MPS I werd voor het eerst ontdekt in de Plott. Beenmergtransplantatie bleek een duidelijke verbetering te geven in de klinische, biochemische en pathologische status van MPS I honden (Breider et al.,1989). Overerving en moleculair defect: MPS I erft over als een autosomaal recessief kenmerk en wordt veroorzaakt door een mutatie in het a-L-iduronidasegen (IDUA). De mutatie bevindt zich niet in het eiwitcoderende deel (exons) van het IDUA maar in de 5' splitsingsplaats tussen intron 1 en exon 2. Introns (= INTergenic RegiONS) worden door een complex mechanisme uit het mRNA geknipt en bevatten normaal geen aminozuurcoderende informatie. De G6A transitie heeft evenwel tot gevolg dat intron 1 tijdens de vorming van het mRNA behouden blijft en aldus een voortijdig stopcodon introduceert, waardoor een veel te korte, niet werkzame polypeptideketen wordt aangemaakt (Menon et al., 1992). Musculaire dystrofie (MD) Klinische symptomen en voorkomen: verschillende X-gebonden musculaire dystrofieën (MD) werden beschreven o.a. in de Golden Retriever en Ierse Terrier. De MD in Golden Retrievers vertoont veel gelijkenissen met Duchenne MD bij de mens (Cooper et al., 1988). Aangetaste pups blijven achter in hun ontwikkeling en de klinische symptomen worden zichtbaar na ongeveer 2 maand. De symptomen evolueren progressief en leiden tot spieratrofie. MD dieren vertonen een verhoogde concentratie kreatinekinase in het serum, hyaline myofibrildegeneratie met mineralizatie, musculaire atrofie en cardiomyopatie (Kornegay et al., 1988). Overerving en moleculair defect: X-gebonden MD in Golden Retrievers erft over als een recessief kenmerk en wordt veroorzaakt door een mutatie in de 3' splitsingsplaats van intron 6 van het dystrophinegen, gelegen op het X-chromosoom. Deze mutatie verandert de normaal aanwezige dinucleotide A-G aan het einde van het intron in het G-G dinucleotide waardoor exon 7 niet als dusdanig wordt herkend en daardoor samen met introns 6 en 7 wordt verwijderd door het splitsingsmechanisme (Sharp et al., 1992). Het dystrophinegen is één van de langste gekende genen, 2000 kb, en het mRNA is 14kb. De mutatie in de Golden Retriever is in positie 736 van het mRNA, dus relatief vooraan. Exon 7 start met de derde base van een codon en eindigt 119 basen verderop met de eerste base van een codon. Indien nu exon 7 wordt verwijderd gaan niet alleen de codons van exon 7 verloren maar tevens wordt er een leesraamverschuiving veroorzaakt waardoor de rest van het mRNA een zinloze inhoud krijgt. In dit speciaal geval wordt tevens een stopcodon gecreeërd zodanig dat een kort transcript ontstaat. Doordat de mutatie overerft als een X-gebonden recessief kenmerk komt de afwijking veel meer tot uiting in mannelijke dieren dan vrouwelijke. Een moleculaire test is beschikbaar waarmee ook vrouwelijke dragers kunnen geďdentificeerd worden. Pyruvaatkinase deficiëntie van erythrocyten (Haemolytische anaemia VIII) Klinische symptomen en voorkomen: pyruvaatkinase (PK) deficiëntie werd beschreven voor de West Highland White Terrier, Basenji, Beagle en de Cairn Terrier. De anemie is meestal ernstig en progressief. Symptomen worden meestal nog gedurende het eerste levensjaar vastgesteld en leiden zonder behandeling tot de dood. Aangetaste dieren zien er doorgaans vrij normaal uit maar vertonen dikwijls een verlaagde activiteit. Ze vertonen miltvergroting, reticulocytose, verkorte levensduur van de rode bloedcellen en erythroďde beenmerg hyperplasie (Searcy et al., 1971). Overerving en moleculair defect: de overerving van deze afwijking gebeurt volgens het patroon van autosomaal recessieve kenmerken. Het pyruvaatkinase aanwezig in erythrocyten (PK1) vormt een tetrameer opgebouwd uit twee ietwat verschillende polypeptiden en is eveneens aanwezig in de lever. Het is een allosterisch enzyme met coöperatieve binding van fosfoënolpyruvaat en gevoeligheid voor fructose-1,6- difosfaat. Het PK1-gen geeft aanleiding tot mRNA van 1629 basen, koderend voor een eiwit van 543 aminozuren. PK1 van lever en erythrocyten verschillen enkel aan het 5' uiteinde en ontstaan wellicht door verschillende processing van hetzelfde mRNA. De mutatie in het PK1-gen heeft tot gevolg dat geen functioneel enzyme wordt aangemaakt (Whitney et al., 1994; Whitney en Lothrop, 1995) en de omzetting fosfoënolpyruvaat naar pyruvaat niet plaatsgrijpt. Hierdoor wordt de omzetting van glucose tot lactaat geblokkeerd met een opstapeling van glycolytische intermediairen en een tekort aan ATP tot gevolg, waardoor de rode bloedcellen uiteindelijk afsterven. Rod/cone dysplasia-1 Klinische symptomen en voorkomen: rod/cone dysplasia-1 werd uitvoerig bestudeerd in de Ierse Setter (Petersen-Jones et al., 1995) en werd eveneens waargenomen in Labrador Retriever, Poedel en Schnauzer. De afwijking, welke gekarakteriseerd wordt door een snel progressief verlies van de fotoreceptors, wordt klinisch ondergebracht bij een groep van aanverwante retinale degeneraties, algemeen aangeduid als progressieve retinale atrofies. In honden met de afwijking lijkt de ontwikkeling van de retina en de fotoreceptors normaal te verlopen tot op een leeftijd van een dertiental dagen, waarna de ontwikkeling van de staafvormige fotoreceptoren stopt. Op een leeftijd van één maand wordt de degeneratie van de staafjes zichtbaar en na vijf maand zijn ze allen gedegenereerd, terwijl dit voor de kegelvormige fotoreceptors op een leeftijd van één jaar het geval is. Biochemisch wordt de afwijking gekarakteriseerd door een accumulatie van cGMP tot meer dan tienmaal het normale niveau en een afwezigheid van cGMP fosfodiesterase activiteit in staafjes (Aguirre et al., 1978). Overerving en moleculair defect: rod/cone dysplasia I erft over als een autosomaal recessief kenmerk. Een mutatie werd gevonden in het gen voor de b-subeenheid van cGMP fosfodiesterase. Deze mutatie, een G6A transitie in codon 807 (exon 21), heeft tot gevolg dat een polypeptideketen wordt aangemaakt die de laatste 49 aminozuren van de normale keten mist. Dit C-terminaal gedeelte is noodzakelijk voor posttranslationele processing en vasthechting aan de membraan (Suber et al., 1993). Tremor (Shaking) Klinische symptomen en voorkomen: tremor werd o.a waargenomen in de Airedale Terrier, Springer Spaniel en Shih tzu en wordt gekenmerkt door het onbedaarlijk trillen van het lichaam, reeds van bij de geboorte (zgn. shaker pups) (Vanvooren, 1995). De afwijking kenmerkt zich door de onderontwikkeling van oligodendrocyten welke in de buurt van de axonen zitten en myeline produceren waardoor een soort overprikkeling ontstaat (Griffiths et al., 1981). Overerving en moleculair defect: tremor erft over als een autosomaal recessief kenmerk en wordt veroorzaakt door een puntmutatie in het gen voor het proteolipide eiwit (PLP). De mutatie onderbreekt de ontwikkeling van de oligodendrocyten (Nadou et al., 1990).
X-linked severe combined immunodeficiency (SCID) Klinische symptomen en voorkomen: SCID is een heterogene groep van genetische defecten die gekenmerkt worden door ernstige abnormaliteiten in zowel de humorale als de cellulaire immuunrespons en die werd waargenomen in o.a. Basset en Beagle. De meest voorkomende vorm van SCID is deze die overerft als een X chromosoom gebonden recessief defect. Het fenotype van X-gebonden SCID bij honden is sterk analoog met X-gebonden SCID bij de mens (Deschęnes et al., 1994). De klinische symptomen omvatten o.a. een verhoogde gevoeligheid voor chronische en dikwijls dodelijke infecties in een vroeg levensstadium. Pathologisch onderzoek heeft aangetoond dat aangetaste dieren een abnormale thymus hebben en een reductie in lymfoďde weefsels, terwijl immunologische studies aantoonden dat het aantal perifere T lymfocyten sterk is afgenomen. X-gebonden SCID is meestal dodelijk rond de leeftijd van vier maanden, tenzij de dieren een beenmergtransplantatie ondergaan (Fisher et al., 1990). Overerving en moleculair defect: genetische studies uitgevoerd op een Basset-familie onthulden dat de mutatie verantwoordelijk voor X-gebonden SCID een 4 baseparen deletie in het gen voor de g-keten van de interleukine 2 (IL-2) receptor betreft. Verwijdering van de 4 nucleotiden veroorzaakt een leesraamverschuiving waardoor een polypeptideketen wordt gemaakt van slechts 21 aminozuren, waarvan er dan nog slechts 10 overeenkomen met die in de normale keten (Henthorn et al., 1994). X-linked Nephritis Klinische symptomen en voorkomen: erfelijke nephritis omvat een groep afwijkingen van het type IV collageen die dikwijls leiden tot slechte nierfunctie. Een karakteristiek morfologisch aspect is het splijten van de glomerulaire basale membraan. X-linked nephritis werd o.a. bestudeerd in de Samojeed (Zheng et al., 1994). Overerving en moleculair defect: ongeveer 85 % van de erfelijke nephritis erft X-gebonden over terwijl de overige 15 % autosomaal dominant of recessief overerft. Type IV collageen moleculen zijn opgebouwd uit drie a-ketens waarvan er zes verschillende types bestaan, a1(IV)-a6(IV). Elke a-keten wordt door een apart gen gecodeerd. Enkel de genen voor de a5(IV) en a6(IV) ketens zijn op het X-chromosoom gelegen. De X-gebonden nephritis wordt veroorzaakt door een transitiemutatie (G6T) in exon 35 van het gen voor de a5(IV) keten. Het codon voor glycine (GGA) is in het mutante allel vervangen door een stopcodon (TGA) waardoor ingekorte, niet functionele a5(IV) ketens worden gevormd (Zheng et al., 1994). GENETISCHE AFWIJKINGEN MET GEKENDE DEFICIENTIE, MAAR VOORLOPIG ZONDER MOLECULAIRE DIAGNOSEMOGELIJKHEID . Deze genetische afwijkingen werden samengebracht in tabel 2.
Albinisme Klinische symptomen: albinisme wordt gekarakteriseerd door afwezigheid van pigment in huid, haar, iris enz., en gaat dikwijls gepaard met een reductie in scherpte van het gezichtsvermogen (Pearson en Usher, 1929; Gwin et al., 1981). Albinisme wordt sporadisch aangetroffen in een groot aantal rassen. Overerving en karakteristieken van de defecte factor: albinisme is een autosomaal recessieve aandoening die wordt gekenmerkt door de afwezigheid van (functioneel) tyrosinase. Tyrosinase is een membraangebonden enzyme dat minstens drie stappen in de omzetting van tyrosine naar melanine katalyseert. Complement C3 deficiëntie Klinische symptomen en voorkomen: C3 deficiëntie werd o.a. vastgesteld in Britse Spaniels. C3 vormt een belangrijk onderdeel van de klassieke complementweg dat geactiveerd wordt door het C3 convertase. Geactiveerd C3 kan binden met vele eiwitten. Deficiëntie kan leiden tot lipodystrofie en mesangiocapillaire glomerulonephritis (Cork et al., 1991).
Overerving en karakteristieken van de defecte factor: complement C3 wordt voornamelijk aangemaakt in de lever en de deficiëntie vertoont een autosomaal recessief overervingspatroon. Het C3 bestaat uit twee ketens, a en b, die covalent met elkaar verbonden zijn door disulfidebruggen. Beide ketens worden gecodeerd door hetzelfde gen. Dit bestaat uit 41 exons en heeft een lengte van ongeveer 40kb. Het gen wordt afgeschreven tot één mRNA dat aanleiding geeft tot één polypeptide dat vervolgens door proteolitische activiteit wordt gesplitst in de a en b ketens (De Bruijn en Fey, 1985).
Canine leukocytenadhesiedeficiëntie (CLAD)
Klinische symptomen en voorkomen: CLAD werd o.a. vastgesteld bij Ierse setters en wordt gekenmerkt door een hoge gevoeligheid aan infecties, gingivitis, slechte wondheling en een zeer hoog aantal leukocyten (Renshaw en Davis, 1979). Overerving en karakteristieken van de defecte factor: CLAD erft over als een autosomaal recessief kenmerk. Bij honden met de deficiëntie werd net als bij het rund (BLAD) een verminderde expressie vastgesteld van CD18, CD11a, CD11b en CD11c op de celmembraan van leukocyten (Trowald-Wigh et al., 1992). In tegenstelling met BLAD waarvoor een mutatie in het CD18-gen werd beschreven, is de mutatie voor CLAD nog niet gedetermineerd. Dermatosparaxie (Ehlers-Danlos syndroom, cutane asthenia) Klinische symptomen en voorkomen: dermatosparaxie is een erfelijke aandoening van de huid die o.a. wordt aangetroffen in de Springer Spaniel, Beagle en Terriers. Het meest uitgesproken klinische symptoom is de kwetsbaarheid van de huid. De geringste schaving veroorzaakt ernstige, gapende snijwonden die evenwel weinig bloeden. De huid van aangetaste dieren vertoont brede, dunne en plooibare littekens. De onderliggende oorzaak is de aanwezigheid van abnormaal type-I collageen in de huid (Hegreberg, 1975).
Overerving en karakteristieken van de defecte factor: dermatosparaxie erft over als een autosomaal dominant kenmerk en is een voorbeeld van een erfelijke ziekte met genetische heterogeniteit. Type-I collageen is een eiwitproduct van twee genen. Het eiwit is opgebouwd als een 3-voudige helix van twee a-1 ketens en één a-2 keten. Elke keten wordt van het gen afgeschreven als een precursor die terminaal een aantal aminozuren bevat die door specifieke endopeptidasen, procollageen I carboxyproteďnase (PCP-C-I) en procollageen I amino-proteďnase (PCP-N-I), dienen verwijderd te worden alvorens de type-I collageen triple helix kan gevormd worden. Elk van deze endopeptidasen bestaat uit minstens twee verschillende polypeptideketens zodat bij de vorming van een type-I collageen molecule minstens zes verschillende genen betrokken zijn. Er kunnen mutaties voorkomen in elk van deze genen. Indien mutatie optreedt in één van de endopeptidasen worden de collageenprecursoren niet omgezet tot het normale structuurproteďne. Heterozygote dieren zullen geen klinische symptomen ontwikkelen aangezien de helft van de normale concentratie van het endopeptidase voldoende is om de collageenprecursoren om te zetten. Dit defect erft in deze omstandigheden recessief over. Dezelfde abnormale collageenmoleculen ontstaan wanneer er een mutatie optreedt in één van beide a-collageenketens zodanig dat de precursor door de endopeptidasen niet kan omgezet worden. In dit geval zullen heterozygote dieren slechts 50 % normale type-I collageenmoleculen bezitten en wel de klinische symptomen vertonen. In dat geval is het defect dominant. De dominante vorm is o.a. waargenomen in honden, katten, paarden en konijnen terwijl de recessieve vorm tot nu toe enkel in runderen en schapen werd aangetroffen. Factor X deficiëntie (Stuart-Prower factor def.) Klinische symptomen en voorkomen: factor X deficiëntie werd o.a. beschreven in de Cocker Spaniel. De afwijking wordt gekenmerkt door een hoog percentage doodgeboren pups, ernstige problemen bij neonatalen met bloedingen in navelstreng, mond en rectum en heeft dikwijls een dodelijke afloop (Dodds, 1973). Overerving en karakteristieken van de defecte factor: de afwijking erft over als een autosomaal dominant kenmerk met onvolledige penetrantie en is in homozygote toestand meestal dodelijk. Het F10-gen is 25 kb lang en bezit 8 exons. Het mRNA is ongeveer 1500 baseparen lang en heeft een ongewoon 3' uiteinde: dit bestaat slechts uit 3 nucleotiden en het poly(A)-signaal bevindt zich in de coderende sekwentie, één base van het stopcodon verwijderd. Factor X wordt gesynthetiseerd als één keten waarbij de lichte en zware keten door het tripeptide arg-lys-arg van elkaar gescheiden worden. Synthese gebeurt in de lever en staat onder de controle van vitamine K. De mutatie verantwoordelijk voor het defect werd nog niet geďdentificeerd. Factor XI deficiëntie (PTA) Klinische symptomen en voorkomen: factor XI deficiëntie of Plasma Thromboplastia Antecedend (PTA) deficiëntie wordt o.a. aangetroffen in de Springer Spaniel, Grote Pyreneër en Kerry Blue Terrier (Knowler et al., 1994). De klinische symptomen zijn meestal minder uitgesproken dan die van de andere haemofilieën. Hematurie en subcutane hematoma's werden wel in bepaalde gevallen geobserveerd. Bloedingen na chirurgische ingrepen (meestal binnen de 24 uur) zijn niet zelden dodelijk (Dodds, 1976). Overerving en karakteristieken van de defecte factor: PTA erft over als een onvolledig dominant kenmerk. Factor XI behoort tot de serine proteases en wordt gecodeerd door een gen van 23 kb bestaande uit 15 exons. De mutatie verantwoordelijk voor de deficiëntie is nog niet achterhaald. Dieren met PTA hebben minder dan 10 % van de normale hoeveelheid van bloedstollingsfactor XI (Knowler et al., 1994). Factor XII deficiëntie (Hageman factor def.) Klinische symptomen en voorkomen: factor XII def. veroorzaakt weinig uitgesproken klinische symptomen en is eerder zeldzaam (Otto et al., 1991). Overerving en karakteristieken van de defecte factor: dit is een autosomaal dominante afwijking veroorzaakt door een defect in het factor XII (F12) gen. Het F12 gen is 12 kb lang, heeft 14 exons, geen typische TATA en CAAT-sequenties in de promotorregio en vertoont alternatieve transcriptie. Het eiwit is verwant met fibronectine en plasminogeen-aktivator. De onderliggende mutatie is nog niet gekend. Gaucherziekte (glucocerebrosidase def.) Klinische symptomen en voorkomen: de Gaucherziekte is een haemolytische aandoening met miltvergroting, beenderafwijkingen, abnormale huidpigmentatie en miltvergroting en behoort tot een groep van cerebroside lipidoses (Hartley en Blakemore, 1973). Volledig herstel is mogelijk via gentherapie door het inbrengen van een normaal funktioneel gen met een retrovirale vector. Overerving en karakteristieken van de defecte factor: de afwijking erft over als een autosomaal recessief kenmerk. Aan de grondslag ervan ligt een mutatie in het gen voor glucocerebrosidase waardoor de efficiëntie van het enzyme verlaagt en waardoor mogelijk een verandering in de antigenische expressie ervan teweeggebracht wordt (Farrow et al., 1982). Het glucocerebrosidasegen werd op 0,2 cM van het PK1-gen gelokaliseerd zodat PK1 (zie pyruvaatkinase def.) ook als genetische merker voor het volgen van de Gaucherziekte kan gebruikt worden (Glenn et al., 1994). Glycogeenopstapelingsziekten Klinische symptomen en voorkomen: er zijn verschillende glycogeenopstapelings-ziekten. Van twee ervan werd de deficiënte factor geďdentificeerd. In type III is dat amylo-1,6-glucosidase en in type VII (Taruiziekte) 6-fosfofructo-1-kinase (Harvey et al., 1990). De ziekte manifesteert zich gewoonlijk in pups van 6 ŕ 12 weken en wordt gekenmerkt door musculaire tremor, vertigo, oogretractie, een verlaging van de lichaamstemperatuur en hyperglucosemie. De dieren vallen soms in coma. De hepatocyten zijn vergroot en buitensporige glycogeenafzetting in het myocard werd waargenomen. Taruiziekte wordt gekenmerkt door spierkrampen en myoglobinuria bij zware inspanning. Glycogeenopstapelingsziekten werden vastgesteld in een aantal gemengde rassen.
Overerving en karakteristieken van de defecte factor: fosfofructokinase katalyseert de omzetting van fructose-6-fosfaat tot fructose 1,6 bifosfaat. Het is een isozyme bestaande uit drie subunits die Muscle (M), Liver (L) en Platelet (L) worden genoemd en die elk door een apart gen worden gecodeerd. Deze genen zijn op verschillende chromosomen gelegen. De L en P subeenheden komen niet tot expressie in de spieren, terwijl de L subunit de hoofdvorm is in de lever. In erythrocyten vormen de L en M subunits tetrameren met een willekeurige samenstelling van beiden. In de hersenen van honden met de Taruiziekte werd een afgeknotte vorm van de M-subunit aangetroffen met afwijkende kinetische eigenschappen (Mhaskar et al., 1991; 1992). GM1 gangliosidosis Klinische symptomen en voorkomen: GM1 gangliosidosis wordt gekarakteriseerd door een degeneratie van het zenuwstelsel, accumulatie van ganglioside in neuronen, hepatocyten en nog enkele andere celtypes en geeft dikwijls aanleiding tot misvormingen van het skelet en werd o.a. vastgesteld in de Engelse Setter en Duitse kortharige Pointer (Alroy et al., 1992). Herstel is mogelijk via beenmergtranplantatie (Obrien et al., 1990). Overerving en karakteristieken van de defecte factor: GM1 gangliosidosis wordt wellicht veroorzaakt door een defect in het gen voor b-galactosidase of eventueel door een mutatie in het gen voor het zogenaamde "protective protein" en erft autosomaal recessief over. Het b-galactosidase splitst de terminale galactose van de oligosaccharide groep van GM1 en breekt de mucopolysaccharide af. Het protective protein zorgt voor de stabiliteit van het b-galactosidase door het te aggregeren in multimeren (Fisher en Alroy, 1992).
GM2 gangliosidosis Klinische symptomen en voorkomen: GM2 werd o.a. waargenomen in de Duitse kortharige Pointer, Beagle, Cairn Terrier, West Highland White Terrier en Poedel. Aangetaste dieren vertonen een nerveus gedrag, verlaagde respons op trainingen, en na 9 tot 12 maand ataxia. De dieren worden dikwijls blind en doof en sterven meestal na een tweetal jaren. De neuronale afzetting van GM2 ganglioside in alle neuronen met granulair materiaal in het cytoplasma leidt tot celvergroting (Karbe, 1973). Overerving en karakteristieken van de defecte factor: de afwijking, die autosomaal recessief overerft, is te wijten aan een deficiëntie van hexosaminidase A. Hexosaminidasen hydrolyzeren de glycosideverbindingen van hexosaminen en N-acetylhexosaminen. Hexosominidase type A werkt in op glycolipiden en beschermt de zenuwcellen tegen de opstapeling van GM2 ganglioside. Haemofilie A Klinische symptomen en voorkomen: haemofilie A is de meest voorkomende vorm van haemofilie bij de hond en wordt veroorzaakt door een defect in coagulatiefactor VIII. Haemofilie A werd in veel verschillende rassen waargenomen waaronder de Ierse Setter, Engelse Setter, Labrador, Duitse Herder, Beagle, Weimaraner, Chihuahua, Collie, Samojeed en Franse Bulldog. De ziekte manifesteert zich gewoonlijk in pups na zes weken tot enkele maanden. De dieren ontwikkelen dikwijls unilaterale of bilaterale gewrichtsvergroting tengevolge hemorrhagia en hemarthrose. Bloedingen in het gastrointestinale kanaal en andere weefsels komen geregeld voor. De ziekte gaat dikwijls gepaard met anorexia (Dodds, 1974).
Overerving en karakteristieken van de defecte factor: het gen voor factor VIII is op het X-chromosoom gelegen en de afwijking erft als een recessief kenmerk over. Tengevolge hiervan wordt de ziekte het meest waargenomen bij mannelijke dieren. Factor VIII is nodig als cofactor, samen met andere factoren en calcium, om factor X te activeren en de intrinsieke prothrombine activator te vormen. Dieren met haemofilie A hebben slechts 1 tot 5 %, en dragers (altijd vrouwelijk) slechts 40 tot 60 % van de normale factor VIII hoeveelheid (Brinkhous et al., 1973). Uitzonderlijk werd een vrouwelijk dier beschreven met haemofilie A, dat dus homozygoot is voor de mutatie (Murtaugh en Dodds, 1988). Hypofibrinogenaemia (fibrinogeen def., factor I def.) Klinische symptomen en voorkomen: hypofibrinogenaemia werd vastgesteld in o.a. Collies en Sint-Bernards en is te wijten aan een defect in coagulatiefactor I (fibrinogeen). De ziekte wordt gekenmerkt door ernstige bloedingen die meestal tot de dood leiden kort na de geboorte. De grootste problemen zijn hemarthrose en bloedingen in de muceuse membranen en subcutane weefsels. De heterozygote toestand kan onderkend worden door een lager dan normaal gehalte aan fibrinogeen (Dodds, 1976). Overerving en karakteristieken van de defecte factor: de ziekte erft over als een onvolledig dominant kenmerk. Fibrinogeen wordt door thrombine omgezet tot fibrine, noodzakelijk voor de bloedklontervorming.
Hypoproconvertinaemia (Factor VII def.) Klinische symptomen en voorkomen: hypoproconvertinaemia is een aandoening te wijten aan een defect in coagulatiefactor VII en werd o.a. aangetroffen in de Beagle (Meyers et al., 1972) en de Alaskan Malamute (Dodds, 1974). De klinische symptomen zijn eerder mild en bloedingen zijn schaars. Problemen kunnen optreden bij trauma's of chirurgie. De dieren zijn heel gevoelig voor demodex schurft. Overerving en karakteristieken van de defecte factor: het defect erft over als een autosomaal dominant kenmerk met onvolledige penetrantie (t.t.z dragers van het dominante kenmerk zijn niet steeds aangetast). Wegens de dominante overerving kunnen zowel homozygoten als heterozygoten in een grote mate als dusdanig geďdentificeerd worden en geweerd voor kweekprogramma's. Hypoprothrombinaemia (prothrombine def.) Klinische symptomen en voorkomen: hypoprothrombinaemia is een van de talrijke defecten in de bloedstolling en wordt veroorzaakt door een deficiëntie van coagulatiefactor II of prothrombine (Dodds, 1977). Overerving en karakteristieken van de defecte factor: de afwijking erft over als een onvolledig autosomaal dominant kenmerk. Het gen voor prothrombine werd geďsoleerd en gecloneerd. Het is 21 kb lang en bestaat uit 14 exons (Degen en Davie, 1987). De mutatie verantwoordelijk voor het defect werd nog niet achterhaald. Megaloblastische anaemia I Klinische symptomen en voorkomen: megaloblastische anaemia I, ook aangeduid als intestinale cobalamine malabsorptie, leidt o.a. tot proteďnuria en misvormingen in het urinaire stelsel en is waarschijnlijk te wijten aan een deficiëntie van de receptor voor de cobalaminefactor in de enterocyten van de brush-border membraan van het ileum (Fyfe et al., 1991).
Overerving en karakteristieken van de defecte factor: megaloblastische anaemia I erft over als een autosomaal recessief kenmerk en wordt veroorzaakt door afwezigheid van bovengenoemde receptor of door het onvermogen ervan om cobalamine (vorm van vit. B12) aan het celoppervlak te binden en/of te binden met transcobalamine II.
Mucopolysaccharidosis type VII (Sly syndrome, b-glucuronidase def.) Klinische symptomen en voorkomen: MPS VII werd voor het eerst beschreven in een gekruist ras (Haskins et al., 1984) en wordt gekarakteriseerd door hepatosplenomegalie, hernia en vertroebeling van de cornea. Soms treden ook neurologische abnormaliteiten op. MPS VII komt veel minder voor dan MPS I en werd voornamelijk in de Beagle aangetroffen (Haskins et al., 1991). MPS VII wordt veroorzaakt door een defect in het b-glucuronidase. D.m.v. vectorgemedieerde gentransfer met een normaal b-glucuronidasegen van de mens kan de normale werking van lysozomen van fibroblasten van MPS VII aangetaste honden volledig hersteld worden (Wolfe et al., 1990).
Overerving en karakteristieken van de defecte factor: MPS VII erft over als een autosomaal recessief kenmerk. Het b-glucuronidasegen is 21 kb lang en bezit 12 exons. Er werden twee verschillende cDNAs beschreven welke ontstaan door alternatieve splicing en waarbij de kortste exon 6 (153 bp) mist. De mutatie welke aanleiding geeft tot het Sly syndrome is nog niet beschreven (Miller et al., 1990). Sphingomyelinosis (Niemann-Pick dis.) Klinische symptomen en voorkomen: sphingomyelinosis of sphingomyeline lipidosis werd o.a. aangetroffen in de Poedel en wordt gekenmerkt door een opstapeling van sphingomyeline in ganglioncellen van het centraal zenuwstelsel wat leidt tot afsterven van de cel. Andere symptomen zijn hepatosplenomegalie, vertraagde groei en ernstige neurologische stoornissen. De afwijkingen treden vrij vroeg op en leiden gewoonlijk tot een vroege dood (Bundza et al., 1979). Overerving en karakteristieken van de defecte factor: sphingomyelinosis erft als een autosomaal recessief kenmerk over en de defecte factor is het enzym sphingomyelinase dat de splitsing van sphingomyeline in fosforylcholine en ceramide katalyseert. Het gen voor sphingomyelinase bestaat uit 6 exons. Testicular feminization (androgeen receptor def.) Klinische symptomen en voorkomen: dit is een defect waarbij een mannelijk dier uiterlijk vrouwelijke geslachtskenmerken ontwikkelt maar toch een normaal mannelijk karyotype (=chromosomenpatroon) vertoont. De afwijking wordt sporadisch aangetroffen in alle rassen (Meyers-Wallen, 1993).
Overerving en karakteristieken van de defecte factor: dit is een recessief X-gebonden aandoening waarvan het defect te vinden is in het androgeenreceptorgen dat 90 kb lang is en kodeert voor een eiwit van ongeveer 900 aminozuren (Meyers-Wallen, 1993). Het eiwit heeft drie functionele delen: een modulerend deel (exon 1), een DNA-bindend gedeelte (exons 2-3) en een androgeenbindend deel (vijf exons). De bestaande mutatie in de hond is nog niet gekend. In het menselijke AR-gen zijn talrijke mutaties gevonden die ofwel stopcodons veroorzaken ofwel aminozuursubstituties tot gevolg hebben. Von Willebrand Disease Klinische symptomen en voorkomen: de Von Willebrand ziekte wordt gekarakteriseerd door interne bloedingen welke bepaalde gelijkenissen vertonen met de defecten optredend bij haemofilie A, waardoor wel eens een verkeerde diagnose wordt gesteld. VWD in honden wordt vooral aangetroffen in Duitse Herder, Golden Retriever, Miniatuur Schnauzer, Doberman Pinscher en Schotse Terrier. Aangetaste dieren vertonen milde tot ernstige diathese, subcutane hematoma's en verlengde postpartum bloedingen. De pups worden soms doodgeboren of sterven soms kort na de geboorte. De ziekte wordt milder na herhaalde drachten en stijgende ouderdom (Dodds, 1970). Overerving en karakteristieken van de defecte factor: VWD erft onvolledig autosomaal dominant of autosomaal recessief over afhankelijk van het type. De factor betrokken bij de Von Willebrand ziekte is het factor VIII-related protein (VIIIR), ook Van Willebrand factor (VWF) genoemd. VIIIR is een groot glycoproteďne dat het hoofdbestanddeel vormt van het VIII-complex. Het circuleert als een multimeer complex van 850 tot 20000 kDa in het plasma. VIIIR wordt gesynthetiseerd door endotheelcellen, hecht zich vast aan de celwand en vormt vervolgens een brug met circulerende bloedplaatjes. Heel recent werd melding gemaakt van een puntmutatie in het VWD-gen van de Schotse Terrier die aanleiding zou geven tot VWD type 3 (Venja et al., 1996). Deze ernstige vorm erft autosomaal recessief over in Schotse Terriers. In dezelfde studie van een Amerikaanse populatie werd een dragerfrequentie van 9,6 % gevonden. De auteurs geven evenwel geen gedetailleerde gegevens over de aard van de mutatie noch over de diagnostische test waarover sprake.
AFWIJKINGEN MET ERFELIJKE INBRENG WAARVAN DE DEFICIENTE FACTOR(EN) NOG NIET IS BEPAALD In tabel 3 werden een aantal afwijkingen bij elkaar gebracht waarvoor een erfelijke inbreng werd vastgesteld. Deze tabel is zeker niet volledig. Enkel de afwijkingen waarvoor onomstotelijk werd bewezen dat ze een erfelijke inbreng bezitten werden hierin opgenomen samen met deze die vrij regelmatig voorkomen en waarvoor aanwijzingen bestaan dat ze (deels) erfelijk worden bepaald. Een aantal van deze afwijkingen bezitten niet enkel een genetische inbreng, maar ondergaan ook belangrijke invloeden vanuit het milieu (voeding etc.). Het best gekende voorbeeld hiervan is wellicht heupdysplasie waarvan de erfelijkheidsgraad op 35 % wordt geschat (Brass, 1989). In tegenstelling met de afwijkingen samengebracht in tabellen 1 en 2 die, op enkele uitzonderingen na, worden bepaald door een defect in één enkel gen, zijn vele van de afwijkingen in tabel 3 multigenisch, t.t.z. meerdere genen dragen elk in een coöperatieve mate bij tot de ontwikkeling van de afwijking en de ernst ervan. Het multifactorieel karakter van deze afwijkingen bemoeilijkt in aanzienlijke mate de studie van het overervingspatroon en de identificatie van de afzonderlijke factoren.
BESLUIT In 1989 werd de eerste erfelijke aandoening bij de hond, haemofilie B, met moleculair genetische technieken opgehelderd (Evans et al., 1989). Sindsdien is het onderzoek van genetische defecten bij de hond in een stroomversnelling gekomen, wat de laatste jaren voor een tiental afwijkingen heeft geresulteerd in de identificatie van de verantwoordelijke mutatie. Voor elk van deze afwijkingen is een moleculair diagnostische test ontwikkeld waarmee zowel aangetaste als dragerdieren kunnen geďdentificeerd worden. De dieren dienen hiervoor niet al te zeer verstoord te worden: een druppeltje bloed, een huidbiopsie, mondswaps etc. kunnen als uitgangsmateriaal gebruikt worden voor de moleculaire test. Verwacht kan worden dat binnen afzienbare tijd ook voor de meeste afwijkingen in tabel 2 de mutatie zal geďdentificeerd worden. De beschikbaarheid van de humane genen betekent dat de corresponderende genen van de hond (en andere huisdieren) snel kunnen geďsoleerd en bestudeerd worden. Compiled and © Ir. F.L. van Tatenhove, reprint omly with permission of the authors and website owner. |
![]() Top
Top
Literature list on request.
![]()
![]()
![]()
![]()
copyright,legal and email statement